Hi, I'm Luca Giudice.
Postdoctoral researcher with expertise in bioinformatics, data science, network science, and AI. Passionate about solving real-world biological problems. Multiple internships in important labs around the world, combined with partnerships involving clinicians, biologists, and medical geneticists
About
I am a Postdoctoral Researcher with expertise in bioinformatics, data science, network science, and artificial intelligence. As the first student in Italy to complete an entire academic career in Bioinformatics/Eng, I have experience solving real-world biological problems, collaborating with experts in various scientific fields, and self-learning mathematical and biomedical concepts. My passion lies in the patient similarity network paradigm and the concept of determining a patient's disease risk based on their genetic similarities with previously known cases. In my work, I am accountable as a project developer, supervisor, and team player.
Experience

- Independent Software Engineer: I have proven myself capable of independently researching, designing, and developing novel machine learning methods for biological applications. My programming skills encompass Python, R, NetworkX, igraph, PyTorch, DALEX, and more.
- Bioinformatician: I have actively investigated the role of coding and non-coding RNAs in brain diseases such as Alzheimer's, Ischemia, and Stroke. For example, I contributed to determining the cirs-7, miR-7, and Cyrano network, a finding published in Cell Reports. As a skilled data scientist and bioinformatics expert, I have successfully analyzed and integrated countless multi-omics sequencing data and multi-modal clinical data.
- Project Manager: I excel at communicating with students, colleagues, and collaborators. I am a proactive supervisor with a strategic mind, keeping both myself and my colleagues focused on the priorities essential for project success. I am very well organized and capable of exploiting managing software like Notion and Obsidian.
- Teacher: Over the 5 years in my lab, I did multiple presentations to introduce and teach about artificial intelligence methods that can be applied to health data. I successfully presented fundamental computational concepts both at simple and advanced levels, such as principal component analysis, clustering algorithms (e.g., k-means), random forests, and neural networks.

- Goal: Unravelling the molecular pathogenesis of combined neuroendocrine cancers by analyzing sequencing data of patients with Combined Large Cell Neuroendocrine Carcinomas (CoLCNEC).
- Data Science: My role was to discover biological markers that can differentiate and classify the 7 histological groups of patients. I encountered the challenge of combining data from various sources while maintaining the true biological signals of patients. To overcome this challenge, I utilized techniques such as batch correction, data normalization, unsupervised machine learning and pathway-based patient classification.
- Award: I was awarded the grant AdR3826/21 named "Neuroendocrine Carcinomas: towards a molecular classification of the disease subtypes for precision medicine".
- My PhD goal was to develop a software for the patient classification that could integrate multiple biological omics data and reveal disease characteristics. I was responsible for designing the project from its feasibility study to its publication as a scientific article. I mastered advanced computer science topics as reproducibility, real-world validation and interpretability of machine learning systems, dockerization, chunking and parallel computing in R and Python.

- Goal: The in-vitro protocol for identifying the involvement of cell-type specific non-coding regulatory elements in gene expression and deregulation is costly and can only detect direct associations.
- Computer Science: I developed a classifier combining a network-based propagation algorithm and a random forest algorithm that can predict the activity of both direct and indirect non-coding regions based on the gene expression of a cell. I learned that time management is a critical factor in the development of a project, and tasks can vary in their importance depending on when they are performed. A project requires two individuals: one who is optimistic and pushes forward, and one who is realistic and focuses on completion.

- Computer Science: I developed a workflow to uncover novel lncRNAs about canine B cell lymphomas. The workflow allowed us to characterize the molecular mechanisms and promote potential prognostic biomarkers about DLBCL, MZL and FL lymphoma.

- Data Analysis: I did an exhaustive characterization of two indolent B-cell lymphomas based on a multi-omics analysis and integration. I used both standard bioinformatic tools and machine learning algorithms.

- Machine Learning: I developed a software module of a patient classifier (netDx) to integrate somatic mutation information to other biological omics (e.g., gene expression, methylation) using graph theory.
Projects
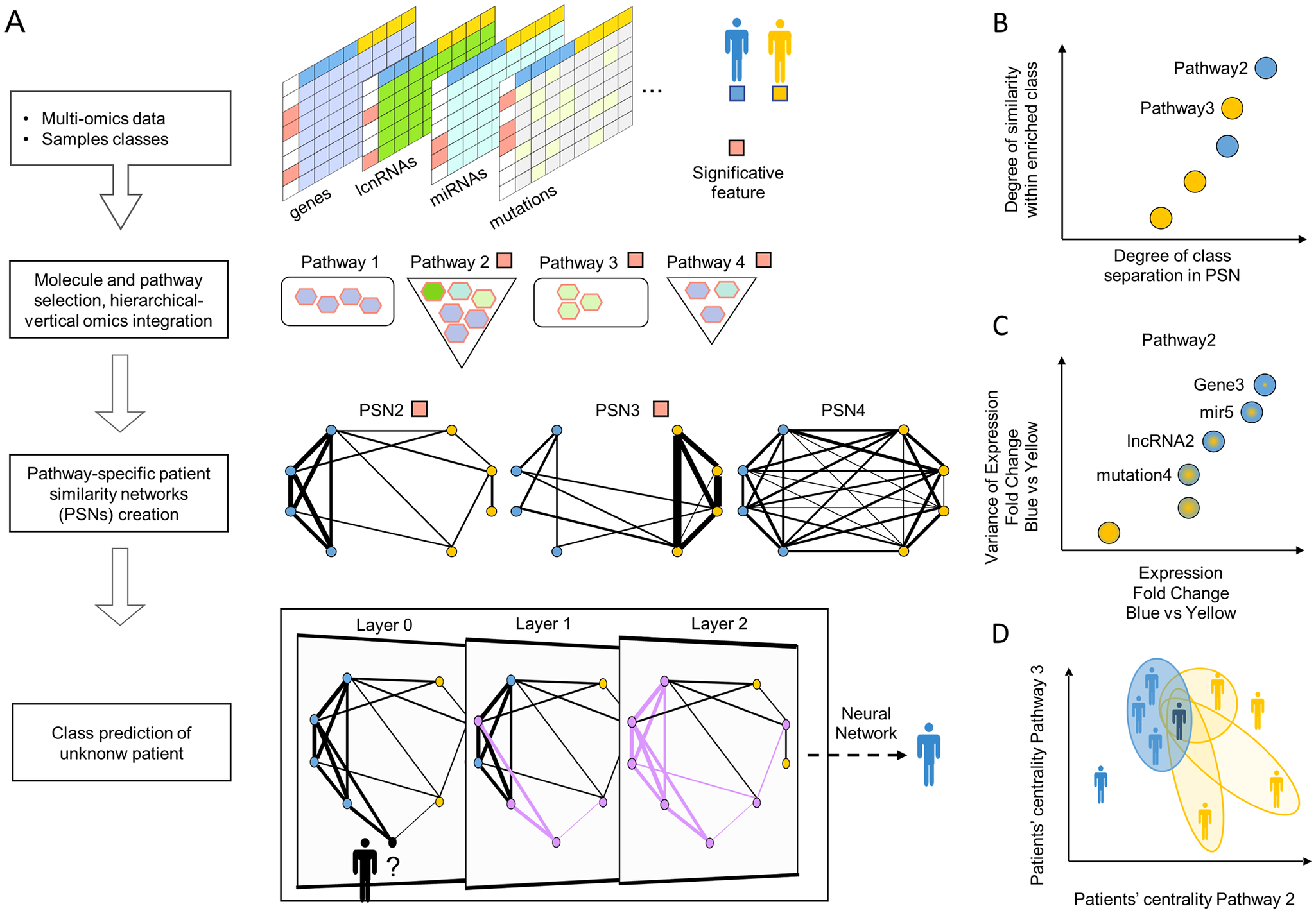
A deep learning patient classifier based on the patient similarity network paradigm
-
Leverages state-of-the-art bioinformatics techniques to select biologically meaningful molecular and pathway features,
the pathway space to integrate different omics and increase the software interpretability,
the PSN paradigm to enable analysis and stratification of patients based on their pathway activity,
and an artificial neural network to account for the similarity information and classify.
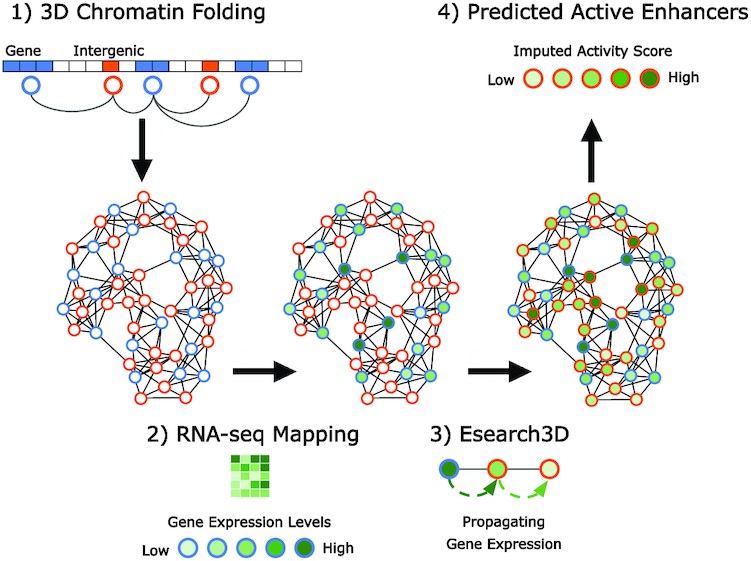
Esearch3D is an unsupervised algorithm to predict enhancers.
-
It reverses engineering the flow of information and identifies intergenic regulatory enhancers using solely gene expression and 3D genomic data.
It models chromosome conformation capture (3C) data as chromatin interaction network (CIN) and then exploits graph-theory algorithms to integrate RNA-seq data
to calculate an imputed activity score (IAS) for intergenic regions.
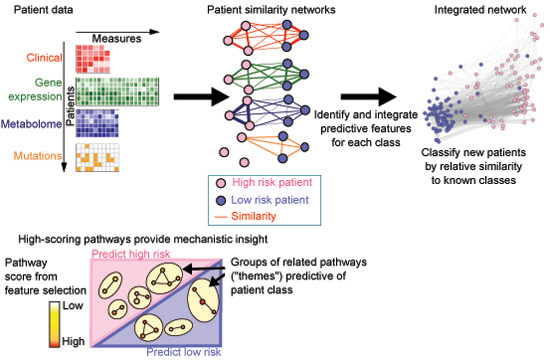
A novel workflow for pathway-based patient classification from sparse genetic data.
-
netDx is for biomedical researchers who want to integrate multi-modal patient data to predict outcome or patient subtype.
netDx builds interpretable machine-learning patient classifiers. Unlike standard machine-learning tools, netDx allows modeling of
user-defined biological groups as input features; examples include pathways and co-regulated elements. In addition to patient classification,
top-scoring features provide mechanistic insight, helping drive hypothesis generation for downstream experiments. netDx currently provides native support
for pathway-level features but can be generalized to any user-defined data type and grouping.
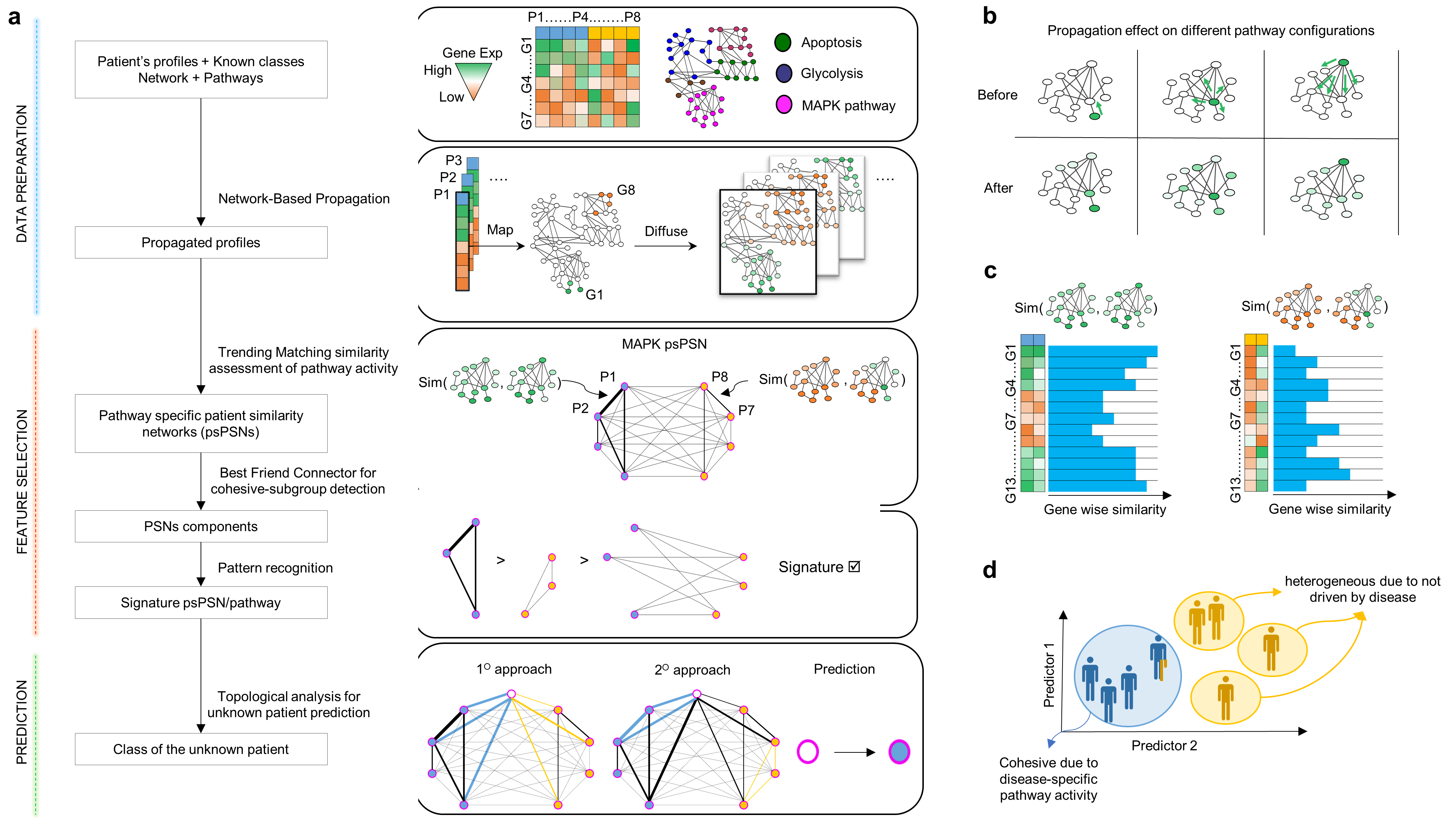
Pathway-based patient classifier uses network science for multi-omics integration and class prediction.
-
We propose Simpati, an innovative and interpretable patient classifier based on pathway-specific patient similarity networks.
The first classifier to adopt ad-hoc novel algorithms for such graph type. It standardizes the biological high-throughput dataset of patient's
profiles with a propagation algorithm that considers the interconnected nature of the cell’s molecules for inferring a new activity score.
This allows Simpati to classify with dense, sparse, and non-homogenous omic data. Simpati organizes patient’s molecules in pathways
represented by patient similarity networks for being interpretable, handling missing data and preserving the patient privacy.
A network represents patients as nodes and a novel similarity measure determines how much every pair act co-ordinately in a pathway.
Simpati detects signature biological processes based on how much the topological properties of the related networks separate the patient classes.
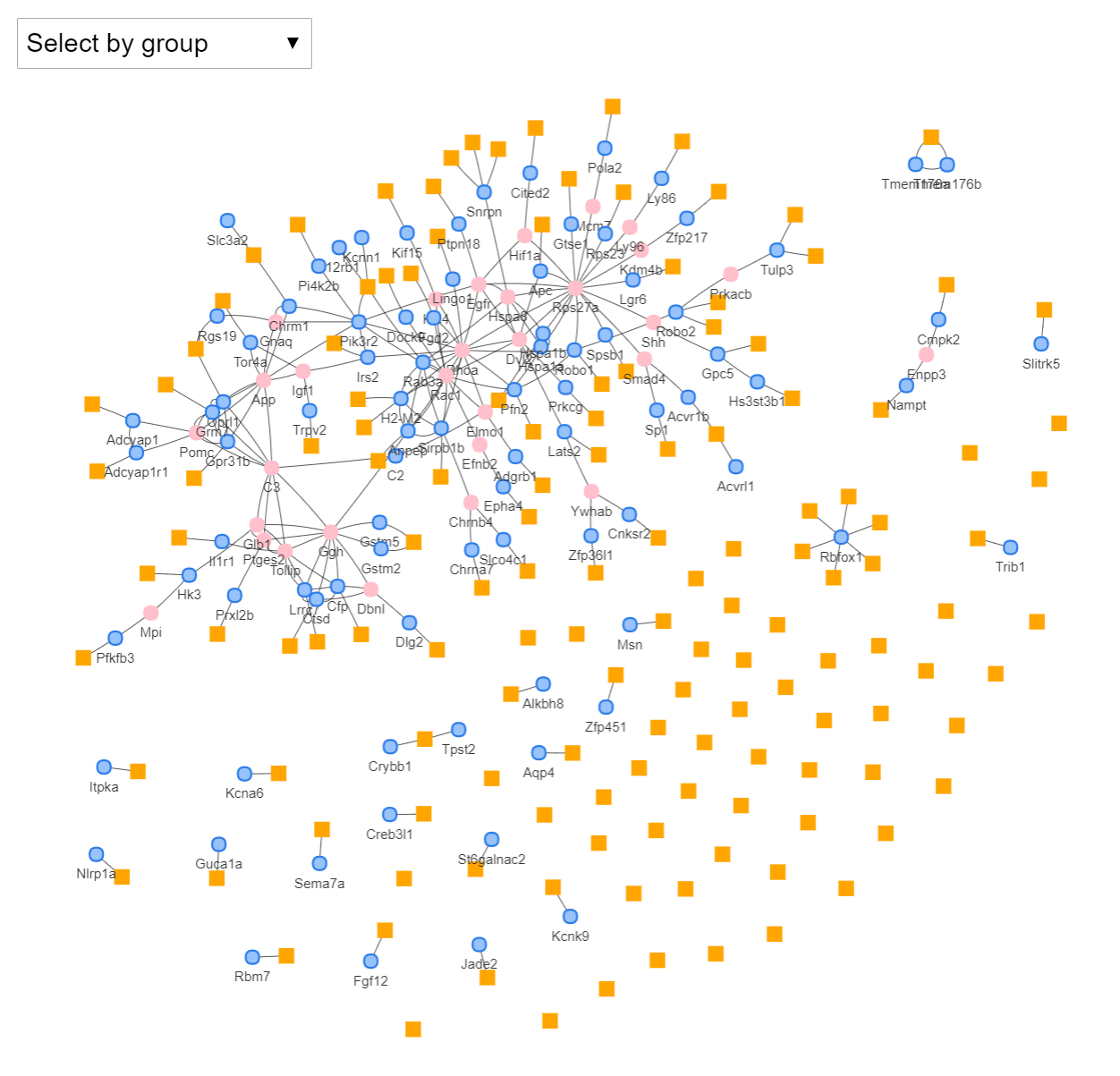
LErNet predicts the function of IncRNAs.
-
LErNet is a method to in silico define and predict the roles of IncRNAs. The core of the approach is a network expansion algorithm which enriches the genomic context of IncRNAs.
The context is built by integrating the genes encoding proteins that are found next to the non-coding elements both at genomic and system level.
The pipeline is particularly useful in situations where the functions of discovered IncRNAs are not yet known. The results show both the outperformance of LErNet compared
to enrichment approaches in literature and its robustness in case of partially missing context information.
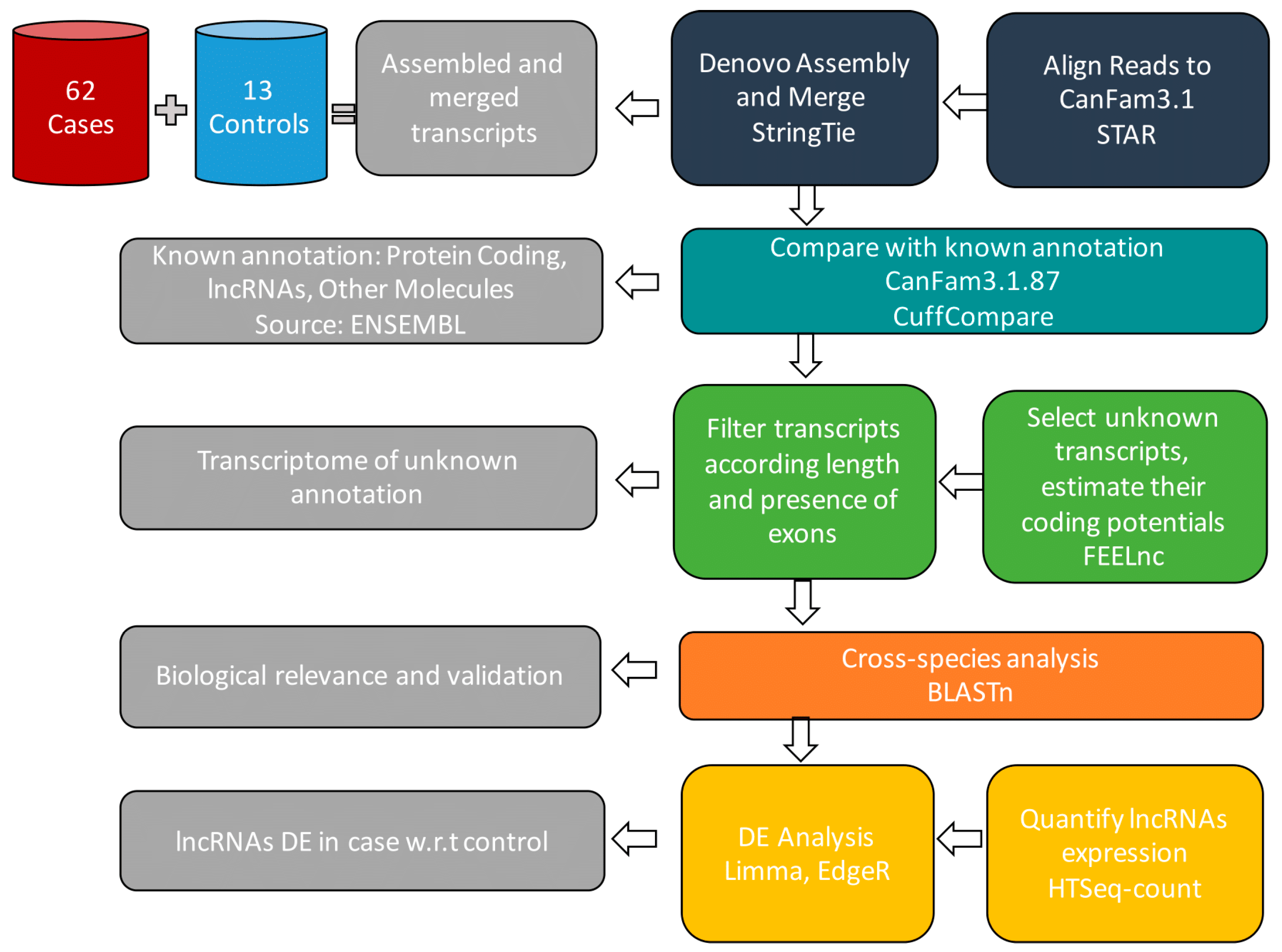
R workflow performing de-novo transcriptome assembly to uncover novel lncRNAs
-
We developed a customized R pipeline performing a transcriptome assembly by multiple algorithms to uncover novel lncRNAs,
and delineate genome-wide expression of unannotated and annotated lncRNAs. Our pipeline also included a new package for high performance system biology analysis,
which detects high-scoring network biological neighborhoods to identify functional modules.
Skills
Languages and Databases
 R
R
Libraries
Frameworks
 Bioconductor
Bioconductor
Other
 Git
Git
 AWS
AWS
 Docker
Docker
Activities

I provided a four-day bioinformatics course invited by Dr. Shen Li at Beijing Shijitan Hospital, Capital Medical University, in China
-
In October 2024, I conducted a four-day (32-hour) bioinformatics course in Capital Medical University (CMU) - Beijing, a culmination of my experience in both bioinformatics research and education.
A surprising observation is the limited understanding of computer science fundamentals underlying many methods applied to health data. This knowledge gap often leads scientists to misapply methods and misuse terminology. For example, Principal Component Analysis (PCA) is frequently used as a visualization technique instead of Multi-dimensional Scaling (MDS). Similarly, terms like correlation, integration, and concatenation are often confused and misused.
Based on my understanding of the critical need for advanced training in the analysis of biological data, I designed this course for both bioinformaticians and computer scientists.


I was delighted to present my review about multi-omics data integration methods at the Finnish Nordic Computational Biology Conference 2024
-
I was delighted to present at the Finnish Nordic Computational Biology Conference 2024 on the patient similarity network paradigm for patient classification. My talk covered the paradigm itself, along with existing methods like netDx (Shraddha Pai et al.) and SUPREME (Ziynet Nesibe Kesimoglu et al.), and introduced my own novel approach, StellarPath. I hope this paradigm gains wider adoption for omics data analysis. My sincere thanks to Ahmed Mohamed and the Nordic Computational Biology group for organizing the conference and giving me the opportunity to speak. An extra thanks also go to my PI Tarja Malm who gives me the chance to grow as researcher and partecipate in these amazing events.

I presented and organized the Finnish Nordic Computational Biology Conference 2023 in Kuopio
-
The Finnish Symposium on Computational Biology 2023 is organised by Luca Giudice under Regional Student Group (RSG) under the International Society for Computational Biology (ISCB) for Finland.
The symposium is tailored for Bioinformaticians and Computational Biologists. It's also an excellent opportunity for researchers or students working with biological data who are keen on exploring new bioinformatics methods, workflows, or challenges.
Education
Verona, Italy
Degree: PhD in Computer Science
Thesis: Artificial Intelligence Techniques Integrate Biological Omics into Graphs for the Prediction and Pathway Analysis of Patient’s Disease
Date: May 2022
University of Verona, Verona, Italy
University of Verona, Verona, Italy
Degree: Master's degree in Medical Bioinformatics
GPA: 4.0/4.0
Date: August 2018
- Data Structures and Algorithms
- Database Management Systems
- Operating Systems
- Machine Learning
- Bioinformatics
Relevant Courseworks: